► Job submission
After verifying the CAPTCHA, a user can upload multiple files or multiple FASTA sequences in a single file for the analysis. To submit a job in the AutomAted RepeaT Identifier, upload a file (with ".fasta/ .fna/ .fa" extension) containing at least two nucleotide sequences in FASTA format. The first line in a FASTA file starts with a ">" (greater-than) symbol followed by accession number and other details. In AARTI pipeline, first word upto the space in FASTA header line will be used to represent the sequence/ organism. Here, the accession number (e.g. NC_022714.1) is used to represent the sequence. Information from FASTA sequence header, which must be in the format shown in Fig. 1 will be used otherwise the information available after ">" symbol will be used to represent the submitted FASTA sequences.

A user can also set the parameters for mono-hexa repeat motifs including distance between two microsatellites. Otherwise, the pipeline will be executed with default parameters (Fig. 2).
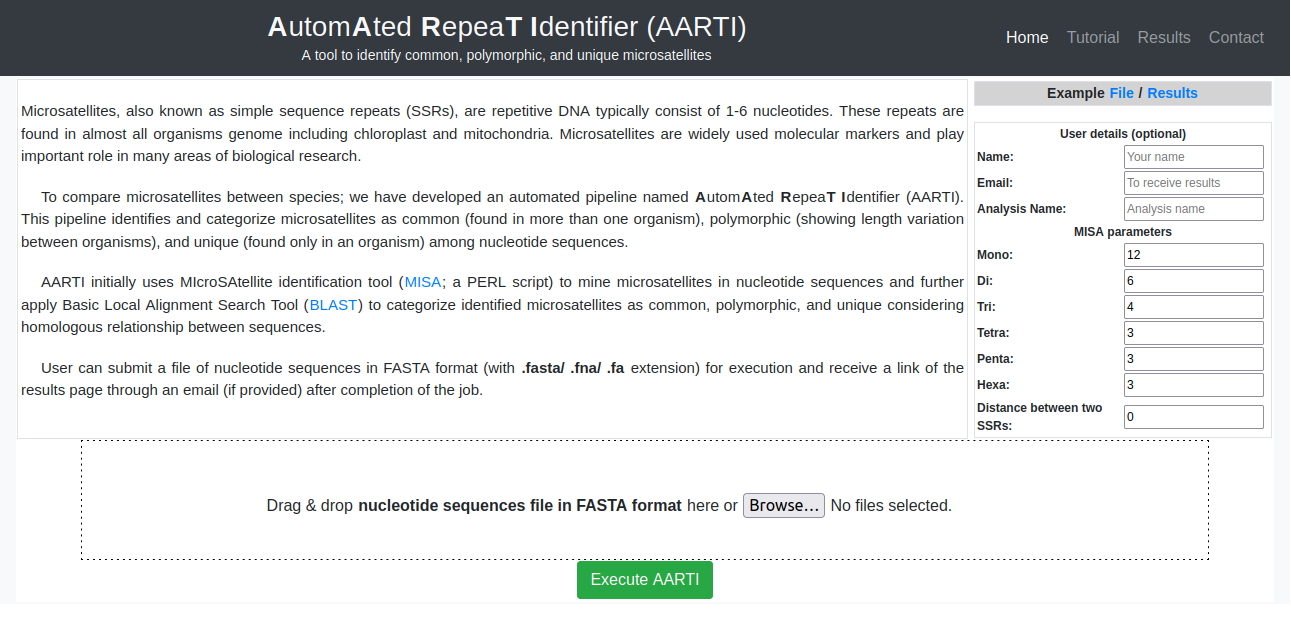
An error message will be displayed, if uploaded nucleotide sequences are not in proper FASTA format or ambiguous characters found in submitted sequences (Fig. 3).
After successful submission a unique job ID will be generated. It will be used to view or download the results available for 10 days since initial job submission (Fig. 4). User may get information of the job on email, if provided during job submission.

► Interpretation of results
Results of two FASTA files (Accession: NC_036024.1 and NC_022714.1), containing complete mitochondrial genome sequences of genus Triticum, are shown. These sequences were downloaded from National Center for Biotechnology Information (NCBI).
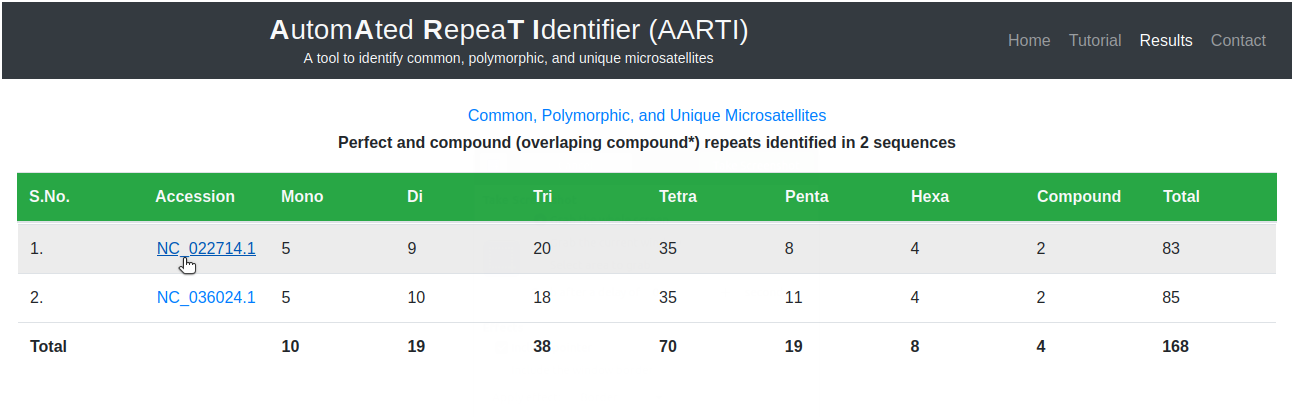
The main results page contains frequency of mono-hexa repeats identified in submitted sequences. The frequency of repeat motifs identified in nucleotide sequences (Fig. 5) can be retrieved by clicking on respective accession number.
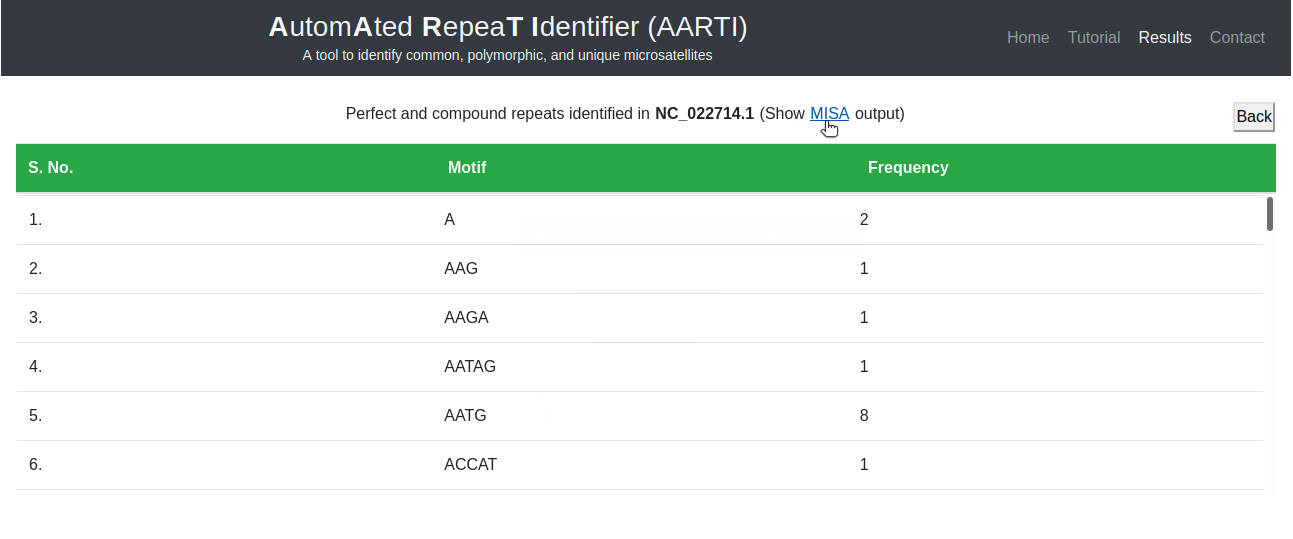
Here user can analyze all repeat motifs identified in selected sequence along with their frequency (Fig. 6).
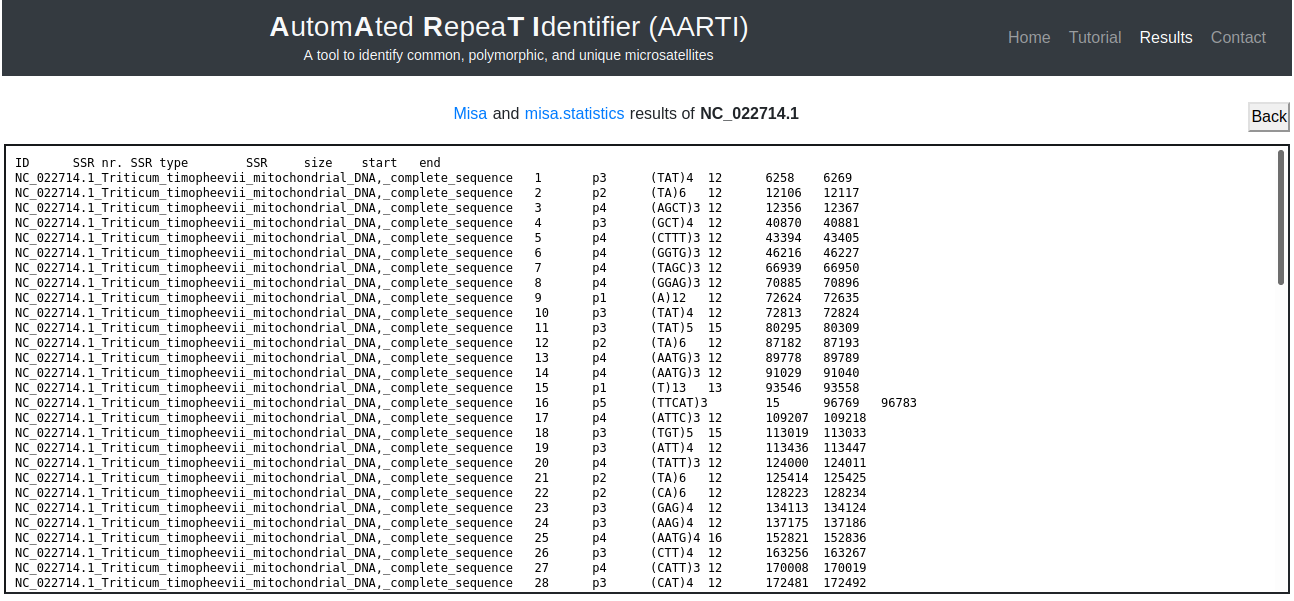
Moreover, MISA results can be retrieved for more details of identified microsatellites (Fig. 7).
► Common, polymorphic, and unique microsatellites
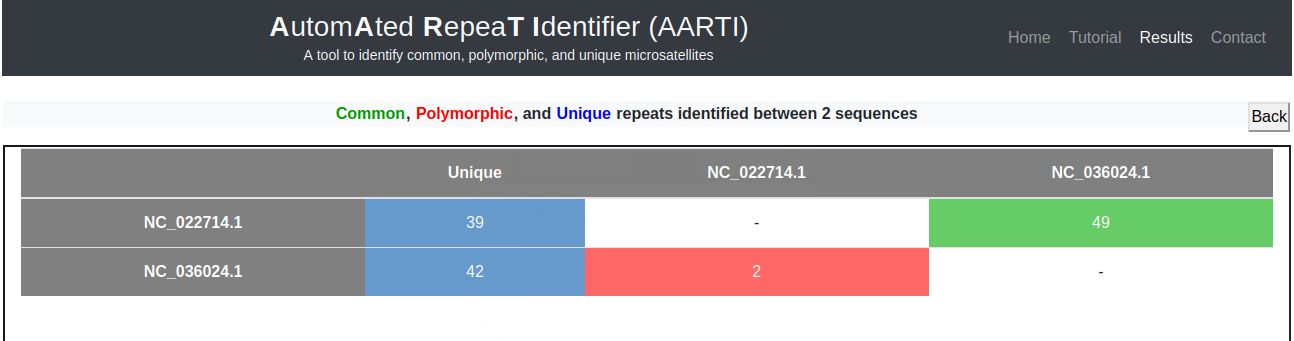
Information of common, polymorphic, and unique microsatellites can be fetched by clicking on the link 'Common, Polymorphic, and Unique Microsatellites' given on the main page (Fig. 5). Identical repeat motifs with equal and varying length, showing significant similarity of flanking sequences (200 nucleotides from both upstream and downstream of microsatellites) on reciprocal BLAST (Basic Local Alignment Search Tool) among the submitted nucleotide sequences, were considered as common and polymorphic microsatellites, respectively. Other repeat motifs and identical repeat motifs showing no significant similarity of flanking sequences were considered as unique microsatellites.
Frequency of common (green) and polymorphic (red) microsatellites identified between each pair of nucleotide sequences appear in the form of a matrix, whereas, total number of unique microsatellites identified in each nucleotide sequence appears in second column (blue) of the matrix (Fig. 8).
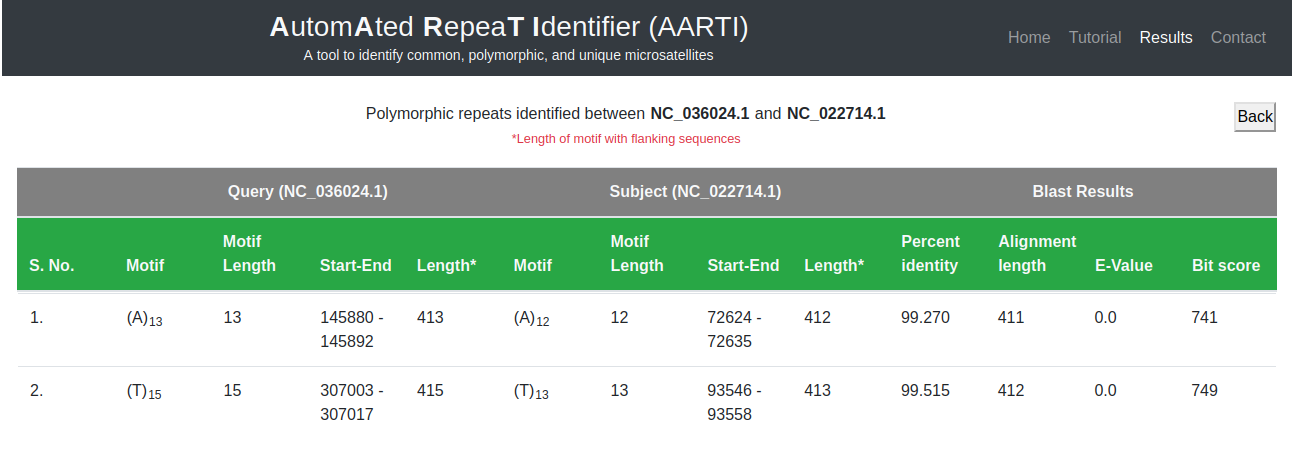
Details of common, polymorphic or unique microsatellites can be fetched by clicking on the respective frequency in the matrix. It will show the details of motif, length, start, and end position along with their sequence alignment results (Fig. 9).
Note: User can download the sample input file or view example results used in this tutorial, which can also be accessed using 'Home' page of AARTI.
***